
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
March 2024
View 2024 - 2021 Archive || 2020 Archive || 2019 Archive ||
2018 Archive || 2017 and Earlier Archive
April 2023:
March 2023:
February 2023:
Glucocorticoid Stress Hormones Stimulate Vesicle-Free Tau Secretion and Spreading in the Braint
The Effects of Insufficient Sleep and Adequate Sleep on Cognitive Function in Healthy Adults
ZCCHC17 Modulates Neuronal RNA Splicing and Supports Cognitive Resilience in Alzheimer's Disease
Effects of Lithium on Serum Brain-Derived Neurotrophic Factor in Alzheimer's Patients with Agitation
2023 Taub Institute Grants for Emerging Research (TIGER) Awardees!
Rie1 and Sgn1 Form an RNA-Binding Complex that Enforces the Meiotic Entry Cell Fate Decision
Memory and Language Cognitive Data Harmonization Across the United States and Mexico
Education as a Moderator of Help Seeking Behavior in Subjective Cognitive Decline
Multicellular Communities are Perturbed in the Aging Human Brain and Alzheimer's Disease
The Neuropathological Landscape of Hispanic and non-Hispanic White Decedents with Alzheimer Disease
The Early-Onset Alzheimer's Disease Whole-Genome Sequencing Project: Study Design and Methodology
Polygenic Risk Score Penetrance & Recurrence Risk in Familial Alzheimer Disease
High School Quality is Associated with Cognition 58 Years Later
Glucocorticoid-Driven Mitochondrial Damage Stimulates Tau Pathology
A Global View of the Genetic Basis of Alzheimer Disease
ARIA in Patients Treated with Lecanemab (BAN2401) in a Phase 2 Study in Early Alzheimer's Disease
Microglia Reactivity Entails Microtubule Remodeling from Acentrosomal to Centrosomal Arrays
Genuine Selective Caspase-2 Inhibition with new Irreversible Small Peptidomimetics
Cell Type-Specific Changes Identified by Single-Cell Transcriptomics in Alzheimer's Disease
Brain Aging Among Racially and Ethnically Diverse Middle-Aged and Older Adults
First Place: Neuroproteasome Localization and Dysfunction Modulate Pathology in Alzheimer's Disease
Clearance of an Amyloid-Like Translational Repressor is Governed by 14-3-3 Proteins
Diet Moderates the Effect of Resting State Functional Connectivity on Cognitive Function
Retromer Deficiency in Tauopathy Models Enhances the Truncation and Toxicity of Tau
Progranulin Mutations in Clinical and Neuropathological Alzheimer's Disease
Wolframin is a Novel Regulator of Tau Pathology and Neurodegeneration
Homotypic Fibrillization of TMEM106B Across Diverse Neurodegenerative Diseases
Correlation of Plasma and Neuroimaging Biomarkers in Alzheimer's Disease
Tubulin Tyrosination Regulates Synaptic Function and is Disrupted in Alzheimer's Disease
The Penalty of Stress - Epichaperomes Negatively Reshaping the Brain in Neurodegenerative Disorders
The Neuronal Retromer can Regulate Both Neuronal and Microglial Phenotypes of Alzheimer's Disease
Deep Learning Improves Utility of Tau PET in the Study of Alzheimer's Disease
Age of Onset of Huntington's Disease in Carriers of Reduced Penetrance Alleles
Caspase-9: A Multimodal Therapeutic Target With Diverse Cellular Expression in Human Disease
Midlife Vascular Factors and Prevalence of Mild Cognitive Impairment in Late-Life in Mexico
The Association Between Sex and Risk of Alzheimer's Disease in Adults with Down Syndrome
Marked Mild Cognitive Deficits in Humanized Mouse Model of Alzheimer's-Type Tau Pathology
Rapid ATF4 Depletion Resets Synaptic Responsiveness after cLTP
Polygenic Risk Score for Alzheimer's Disease in Caribbean Hispanics
Recognition Memory and Divergent Cognitive Profiles in Prodromal Genetic Frontotemporal Dementia
The Microtubule Cytoskeleton at the Synapse & The Synaptic Life of Microtubules
Optimizing Subjective Cognitive Decline to Detect Early Cognitive Dysfunction
The AD Tau Core Spontaneously Self-Assembles and Recruits Full-Length Tau to Filaments
Olfactory Impairment is Related to Tau Pathology and Neuroinflammation in Alzheimer's Disease
Pathogenic Role of Delta 2 Tubulin in Bortezomib-Induced Peripheral Neuropathy
1: Diet, Pace of Biological Aging, and Risk of Dementia in the Framingham Heart Study
2: The Matrix Receptor CD44 Is Present in Astrocytes throughout the Human Central Nervous System and Accumulates in Hypoxia and Seizures
3: Microglia Measured by TSPO PET are Associated with Alzheimer's Disease Pathology and Mediate key Steps in a Disease Progression Model
4: A Comparative Study of Structural Variant Calling in WGS from Alzheimer's Disease Families
2: The Matrix Receptor CD44 Is Present in Astrocytes throughout the Human Central Nervous System and Accumulates in Hypoxia and Seizures
3: Microglia Measured by TSPO PET are Associated with Alzheimer's Disease Pathology and Mediate key Steps in a Disease Progression Model
4: A Comparative Study of Structural Variant Calling in WGS from Alzheimer's Disease Families
Diet, Pace of Biological Aging, and Risk of Dementia in the Framingham Heart Study
 |  | |
| Aline Thomas, PhD | Yian Gu, MD, MS, PhD |
People who eat healthier diets are less likely to develop dementia, but the biological mechanism of this protection is not well understood. Literature also suggests that a healthy diet slows down the processes of biological aging. However, it is unclear whether the slower pace of biological aging is a mechanism linking healthy diet with reduced dementia risk. In our recent study published in Annals of Neurology, we tested the hypothesis that a healthy diet protects against dementia by slowing down the body’s overall pace of biological aging.
In 1,644 participants (≥60 years-old, free of dementia) from the Framingham Offspring Cohort, we assessed healthy diet as long-term adherence to the Mediterranean-Dash Intervention for Neurodegenerative Delay diet (MIND), over 4 visits spanning 1991-2008. Developed in 2015 for prevention of dementia, the MIND diet emphasizes high intake of neuroprotective foods such as fish, green leafy vegetables, berries, and nuts, while minimizing intake of red meat, butter, and sweets. We measured the pace of aging from blood DNA methylation data collected in 2005-2008 using the DunedinPACE epigenetic clock. Developed by studying changes in 19 indicators of the integrity of several different systems in the body (cardiovascular, hepatic, renal, pulmonary, periodontal, immune, metabolic, endocrine), DunedinPACE functions like a speedometer for the aging process, summarizing the overall the rate of change across these systems. Participants were followed for incident dementia from 2005-2008 visit through 2018.
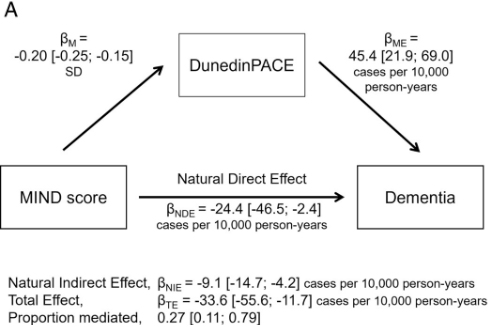
Figure 3A. Mediation analysis of diet association with dementia by the DunedinPACE epigenetic clock measure of the pace of aging.
Our study confirmed that adherence to a healthier diet slowed the pace of aging and reduced risks for dementia and mortality. Slower DunedinPACE accounted for 27% of the diet-dementia association and 57% of the diet-mortality association.
Our findings show that multi-system processes of aging mediate part of the relationship of healthy diet with reduced dementia risk. Advocating healthy diet for the purpose of dementia prevention can now be supported by partially clarified biological mechanisms. Monitoring the pace of aging may inform dementia prevention.
Aline Thomas, PhD
Postdoctoral Research Scientist in the Taub Institute
at3702@cumc.columbia.edu
Yian Gu, MD, MS, PhD
Associate Professor of Neurological Sciences (in Neurology, Epidemiology, the Gertrude H. Sergievsky Center, and the Taub Institute) at CUMC
yg2121@cumc.columbia.edu
 |  | |
| Osama Al Dalahmah, MD, PhD | James E. Goldman, MD, PhD |
CD44 is a transmembrane glycoprotein and a receptor for extracellular matrix, particularly hyaluronan and osteopontin. It has been mainly studied in T cells and in a variety of cancers, in which it promotes an epithelial to mesenchymal transition and metastasis, but much less so in the central nervous system (CNS), where it is expressed in some but not all astrocytes. For example, the so-called protoplasmic astrocytes of the cerebral cortex and other gray matter areas, which carry out critical homeostatic functions for neurons and enwrap synapses, do not express CD44, whereas astrocytes in the white matter and astrocytes with long processes that originate at the surface of the cortex do express this receptor.
In our latest work, recently published in Cells, we performed the first analysis of CD44 in astrocytes over the entire human CNS, finding it in many astrocytes throughout all regions. It is present in astrocytes just under the ependymal lining of the ventricles, some of which cells extend thin processes in between ependymal cells into the ventricle. CD44 astrocytes in the brain stem and spinal cord send processes that wrap around neurons, such as motor neurons. This is never seen in the cortex. Gene expression in these astrocytes is in some ways different from that in protoplasmic astrocytes, and in particular they do not express genes involved in glutamate and potassium homeostasis. Dr. Osama Al Dalahmah and I have used single nuclear RNA sequencing on human brains from the NY Brain Bank, and have found many gene expression differences between CD44 and non-CD44 astrocytes. We also found that protoplasmic astrocytes can convert to a CD44 type of astrocyte in neurological diseases. In this paper and in other studies, we show that protoplasmic astrocytes in Huntington’s Disease and Parkinson’s disease, epilepsy, and conditions that produce ischemia or hypoxia, such as strokes, convert to the CD44 type. Others have found cortical CD44 astrocytes in Alzheimer’s disease and fronto-temporal dementia.
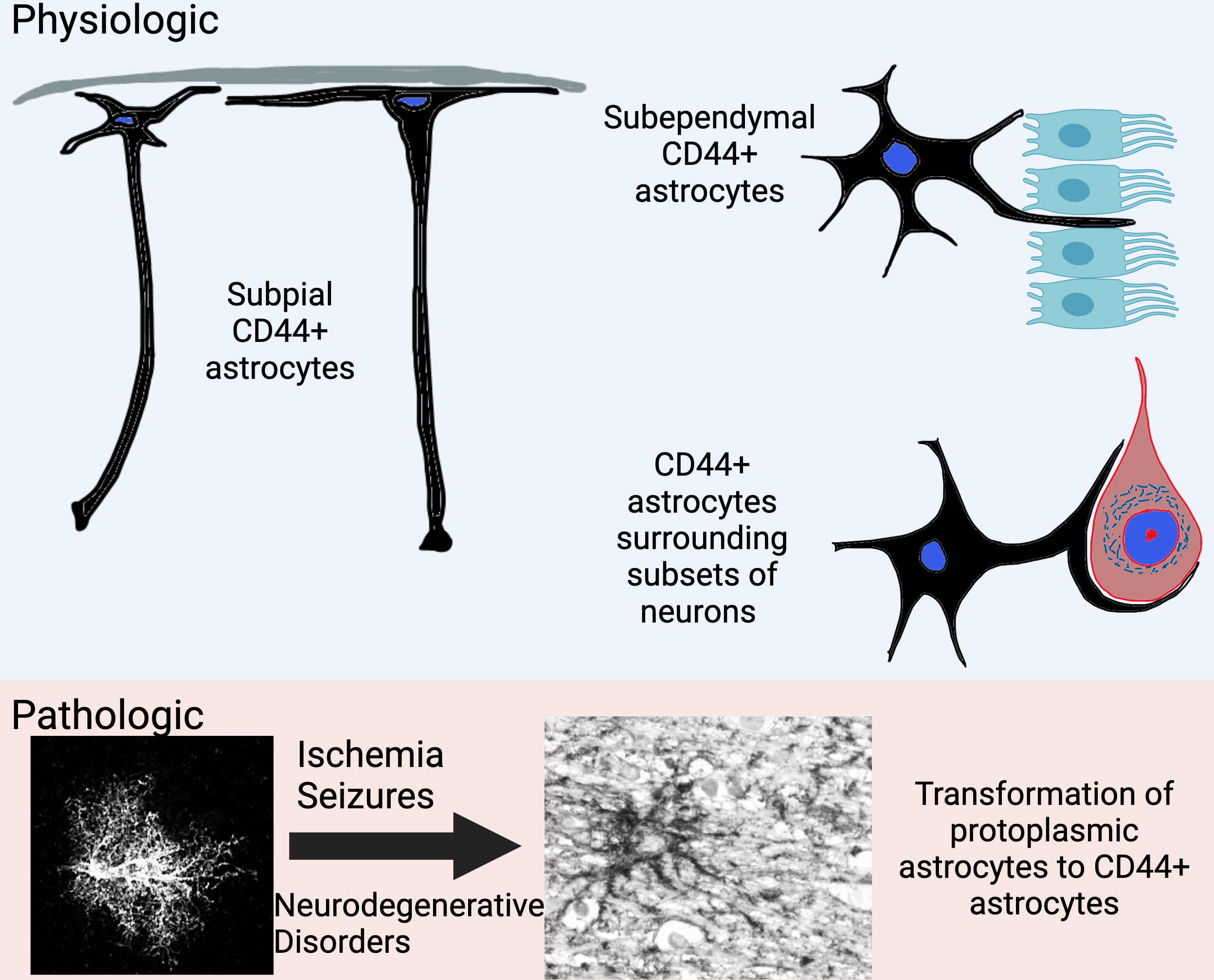
Graphical Abstract
This conversion of protoplasmic to a CD44 type of astrocytes is of great importance for astrocyte function, since during this transition astrocytes downregulate many of the normal protoplasmic genes, including ones that encode glutamate transporter and metabolism proteins. They also change their morphologies, from the bushy types of protoplasmics to ones with longer unbranched processes. In temporal lobe epilepsy, we found that the protoplasmics that convert to the CD44 type enwrap hippocampal pyramidal neurons, a feature never found in the normal brain. We do not know yet how this wrapping might influence neuronal function.
We are now trying to find what signaling through the astrocyte CD44 receptor means to the functions and shapes of astrocytes, and how changes in astrocyte function due to the acquisition of CD44 might influence the functions of neighboring cells, such as neurons. Interestingly, the cytoplasmic domain of CD44 can be cleaved off, after which it enters the nucleus and acts as a transcription factor. This occurs in cancers but has never been studied in astrocytes. We should know soon how gene expression patterns change when astrocytes that do not express CD44 begin to express it.
Osama Al Dalahmah, MD, PhD
Assistant Professor in Pathology and Cell Biology
oa2298@cumc.columbia.edu
James E. Goldman, MD, PhD
Professor of Pathology and Cell Biology (in Psychiatry)
jeg5@cumc.columbia.edu
 |  | |
| Samantha Rossano, PhD | Patrick J. Lao, PhD |
With TSPO PET imaging, which targets the translocator protein that is involved in cholesterol transport across the outer mitochondrial membrane within microglia, we can investigate the spatial co-localization of microglia with traditional amyloid, tau, and neurodegeneration imaging measures in individuals across the AD continuum. Microglia are inflammatory cells in the CNS that surveil the brain to clear toxins, among other functions. Microglia clear amyloid-β and neurofibrillary tau pathology as well as neurodegeneration-related products via pathways including complement-mediated phagocytosis. However, there is growing evidence that microglia may potentiate AD pathology. Chronic complement cascades can activate the inflammasome within microglia, affecting their function, leading to a cholesterol transport imbalance (which in turn affects APP processing into amyloid-β), and calcium imbalance (with in turn affects enzymatic/kinase/phosphatase activity on tau). In collaboration with Dr. William Kreisl and first-author Samantha Rossano, we recently reported in Alzheimer’s & Dementia that microglia are elevated in key AD brain regions in relation to amyloid burden, tau propagation across Braak stage regions, and local tau-related neurodegeneration and cognitive impairment in a clinically characterized sample.
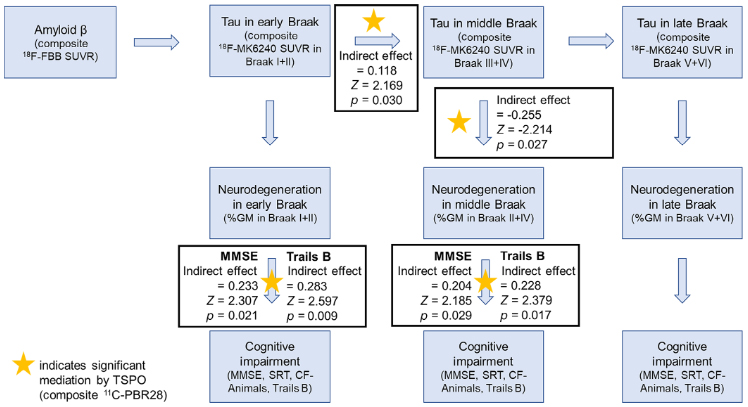
Figure 2. TSPO expression by 11C-PBR28 uptake (SUVR) significantly mediated pathways along AD progression model. SUVR, standardized uptake value ratios; TSPO, translocator protein.
Greater microglia density was associated with amyloid and tau pathology after adjusting for neurodegeneration, which suggests co-localization of microglia and AD pathology that is not due to microglia simply being present in the same region to clear neurodegeneration-related products. Further, microglia density was added as a mediator in an AD progression model, from amyloid to early Braak stage tau, to middle Braak stage tau, to late Braak stage tau, as well as from tau to neurodegeneration to cognition within each Braak stage. Despite direct associations between amyloid, tau, and microglia, microglia did not mediate the association between amyloid and tau. Other work has suggested that astrocytes may be involved in amyloid-related tau accumulation within early Braak stage regions. We found that microglia mediated the association between early Braak stage tau and middle Braak stage tau, suggesting a role in tau propagation across the brain. Microglia also mediated the association between tau and neurodegeneration in middle Braak stage regions and between neurodegeneration and cognition, suggesting a role in tau-related neurodegeneration and cognitive impairment. In our sample, microglia did not mediate associations in late Braak stage regions, potentially due to the restricted clinical range of participants that can participant in PET studies (i.e., individuals with higher CDR and greater tau burden in late Braak stages were excluded).
While many disease modifying treatment strategies focus on amyloid and tau pathology, inflammation may represent another critical disease pathway to target for intervention. Inflammatory pathways may also represent common pathways across different diseases. Our study supports future work that should investigate microglia function above and beyond their spatial localization with AD pathology, as well as cross-talk between other inflammatory cell types in a disease stage and region-specific manner.
Patrick J. Lao, PhD
Assistant Professor of Neurological Sciences (in Neurology and the Gertrude H. Sergievsky Center)
pjl2133@cumc.columbia.edu
A Comparative Study of Structural Variant Calling in WGS from Alzheimer's Disease Families

Badri N. Vardarajan, PhD, MS
The National Institute on Aging Alzheimer’s Disease Sequencing Project (ADSP) was instituted with overarching goals to: 1) identify new genes and genetic variations that contribute to increased risk or protection against Alzheimer’s disease (AD) and related dementias (ADRD); 2) provide insight as to why these genes and variations impact AD/ADRD, and 3) identify potential avenues or approaches to transform genetic results into meaningful therapeutic targets. Taub Institute investigators are an integral part of the ADSP Consortia, and the role of our team is to analyze genetic data to identify risk factors for AD/ADRD. Most of the genetic data available through ADSP are single nucleotide polymorphisms (SNPs) and small insertions/deletions (indels). The sequence analysis protocol for this data does not capture larger structural variation (SVs) in the human genome, because reliable detection of SVs from WGS remains a significant challenge. We hypothesized that large SVs could contribute significantly to the risk of AD/ADRD, since they affect more genomic region than SNPs. There are many algorithms and software that use information from different aspects of WGS, such as sequencing depth, paired end reads, and mapping quality to compute SVs. However, no single algorithm captures the complete spectrum of structural variation with accuracy in the human genome.
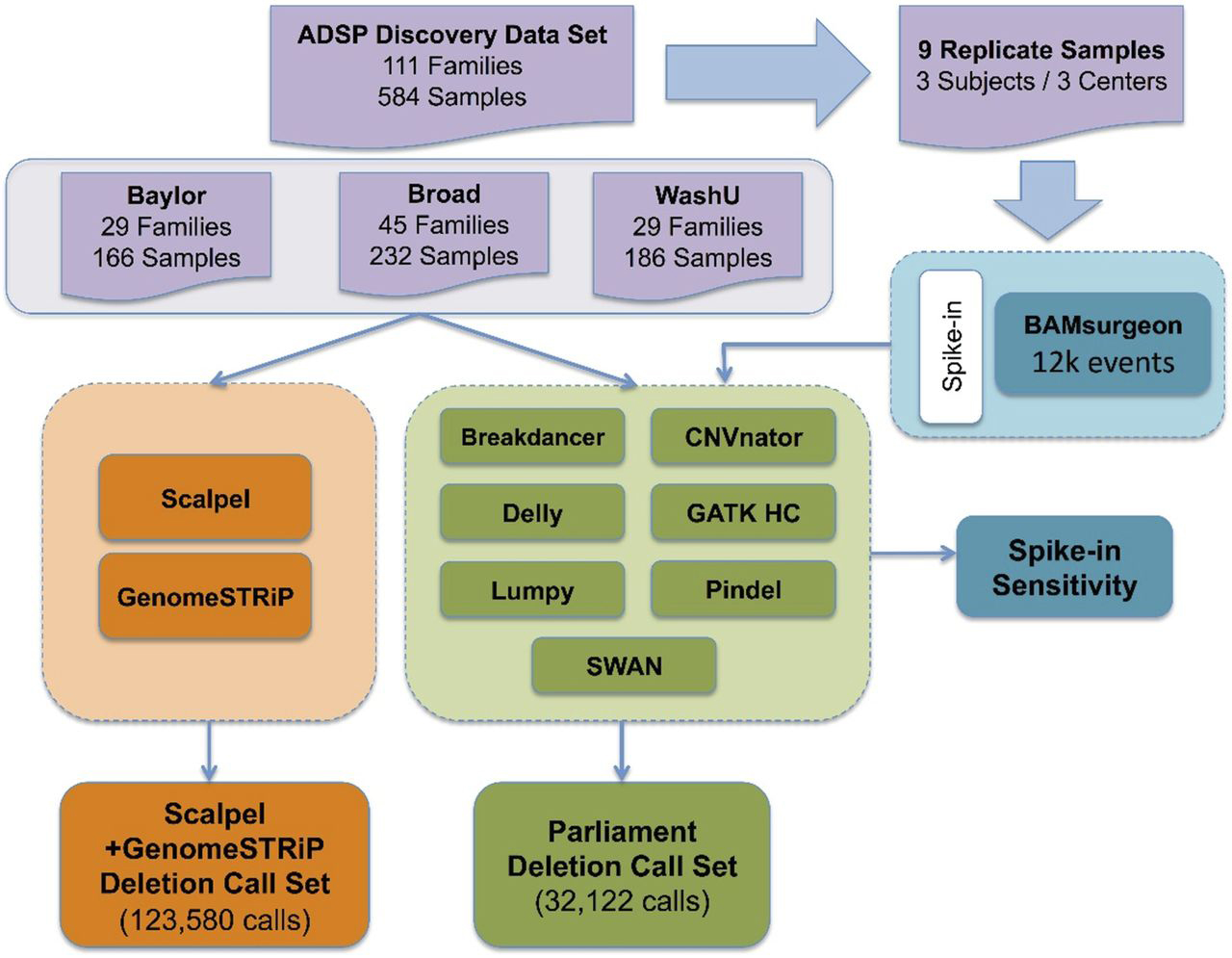
Figure 1. Overview of Alzheimer’s Disease Sequencing Project’s SV/indel calling and analysis pipeline. Two parallel pipelines, Scalpel + GenomeSTRiP (orange) and Parliament (green), were combined to perform SV/indel call merging, QC, genotyping, and reassembly for 584 samples from three sequencing centers. Nine replicated samples were used to measure individual SV/indel caller sensitivity via variant spike-in studies.
To tackle this problem, we created a protocol for variant calling, sensitivity analysis, and laboratory validation for generating a high-quality SV call-set in WGS from the ADSP. This dataset contains 578 individuals from 111 families. We applied two complementary pipelines (Scalpel and Parliament) and several variant callers for SV/indel calling to produce a high-quality annotated call-set. To measure sensitivity, we spiked-in SVs of different sizes in nine sample replicates. We generated a novel metric, D-score, that compares sharing between family members and with unrelated individuals to evaluate specificity of SV calling algorithms. Accuracy of calling was evaluated by Sanger sequencing of predicted loss-of-function (LOF) variants, variants near AD candidate genes, and randomly selected genome-wide deletions between 2 to 17,000 bp.
As recently reported in Life Science Alliance, we identified a total of 152,301 deletions and 78.1% of these variants that were tested in the lab were validated by Sanger sequencing. We found that Scalpel was more accurate in calling deletions <100 bp, and Parliament was optimal for calling deletions >900 bp. We validated 83.0% (88/106) and 72.5% (37/51) of calls made by Scalpel and Parliament, respectively. To summarize, we developed a flexible protocol to generate a high-quality deletion call-set and a truth set of Sanger sequencing validated deletions with precise breakpoints between 1 and 17,000 bp. We are now applying this protocol to large multiplex families from the US and Dominican Republic to identify segregating SVs with AD/ADRD.
Badri N. Vardarajan, PhD, MS
Assistant Professor of Neurological Science (in Neurology, the Sergievsky Center, and the Taub Institute) at CUMC
bnv2103@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.