
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
June 2014
Neurological Disorders: Quality-Control Pathway Unlocked

Asa Abeliovich, MD, PhD
Parkinson's disease, a progressive neurodegenerative disorder, has long been hypothesized to be caused by defects in organelles called mitochondria, which power mammalian cells through the production of ATP molecules. An accumulation of dysfunctional mitochondria may lead not only to a cellular energy crisis, but also to excessive production of toxic by-products. Two enzymes implicated in Parkinson's disease, PINK1 and parkin1, 2, are thought to be involved in the disposal of defective mitochondria, but how the two proteins interact has been unclear. A trio of studies (by Kane et al.3, writing in the Journal of Cell Biology; by Kazlauskaite et al.4, in the Biochemical Journal; and by Koyano et al.5, on page 162 of this issue) now report that phosphorylated ubiquitin protein is the link between PINK1 and parkin, providing insights into a complex system of parkin regulation.
Kinase enzymes such as PINK1 alter the behaviour of target proteins through the addition of phosphate groups, a process called phosphorylation. PINK1 is imported to mitochondria and, in healthy cells, undergoes rapid degradation6. However, if mitochondria are defective or damaged (for example by exposure to CCCP, a poison that blocks ATP production), PINK1 accumulates, becoming anchored to the outer mitochondrial membrane with its kinase domain exposed to the cytoplasm.
Damaged mitochondria also attract parkin, which is otherwise dispersed throughout the cytoplasm in healthy cells7. Parkin is a ubiquitin ligase, which adds ubiquitin proteins (either singly or in polyubiquitin chains) both to itself through autoubiquitination and to nearby target proteins. Ubiquitinated proteins can serve as a signal to the cell that a cellular compartment should be degraded, which in damaged mitochondria leads to their timely disposal7, a process known as mitophagy.
Mutations in either PINK1 or PARKIN that underlie rare familial forms of Parkinson's disease disrupt mitophagy, implicating this cellular pathway in Parkinson's disease7. Furthermore, PINK1 mutations impede the recruitment of parkin to damaged mitochondria, suggesting that the proteins act in a linear pathway. Consistent with a PINK1–parkin quality-control pathway, mutations in pink1 or parkinin fruit flies cause accumulation of defective mitochondria and cellular degeneration8,9.
Initial models proposed that PINK1 phosphorylates and so activates parkin in damaged mitochondria. Although direct phosphorylation of parkin by PINK1 has been documented10, this modification does not seem to be sufficient for full activation of parkin's ubiquitin-ligase activity3, 4, 5, 10. In search of a functional connection between PINK1 and parkin, three groups undertook cell-wide protein analyses and biochemical studies, and found the missing link between the two – phosphorylated ubiquitin (phospho-ubiquitin).
Each study showed that, in cells in which PINK1 was activated by CCCP treatment, PINK1 phosphorylates ubiquitin at a serine amino-acid residue (serine 65). Strikingly, a corresponding serine-65 residue in a ubiquitin-like domain is the aforementioned target of PINK1 phosphorylation on parkin10. Subsequent analyses by all three groups demonstrated that modified ubiquitin, in turn, induces parkin activity (Fig. 1).
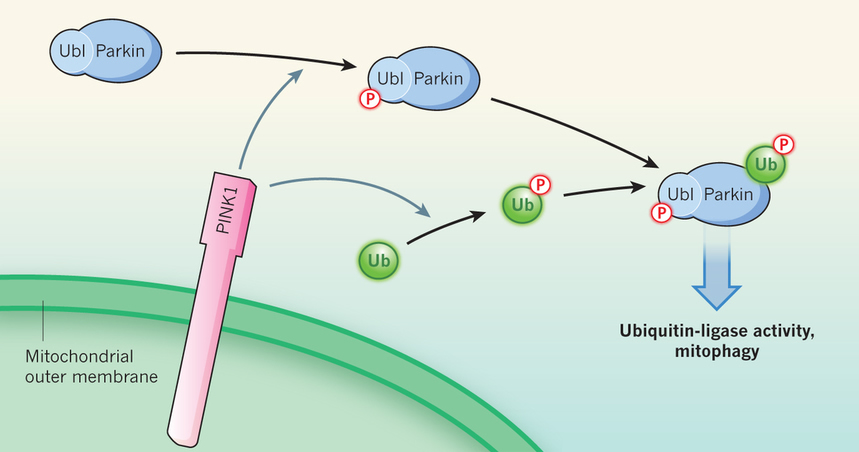 Fig 1: PINK1 and parkin in mitochondrial quality control. Mitochondrial damage leads to anchoring of the PINK1 enzyme to the outer mitochondrial membrane, with its kinase domain facing the cytoplasm. PINK1 adds a phosphate group (P) to the ubiquitin-like domain (Ubl) of the ubiquitin-ligase enzyme parkin. Three studies3, 4, 5 find that PINK1 also phosphorylates the ubiquitin (Ub) protein itself. Phosphorylated ubiquitin directly binds to and activates parkin. Activated parkin ligates ubiquitin and phospho-ubiquitin molecules to nearby target proteins, leading to disposal of the damaged mitochondria through mitophagy. |
[Read the full article in nature]
Asa Abeliovich, MD, PhD
aa900@columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.