
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
June 2017
#1 Post translational Remodeling of Ryanodine Receptor Induces Calcium Leak Leading to Alzheimer's Disease like Pathologies and Cognitive Deficits
#2 Neuropathologic Features of TOMM40 '523 Variant on Late-Life Cognitive Decline
#2 Neuropathologic Features of TOMM40 '523 Variant on Late-Life Cognitive Decline
 |  | ||
| Andrew R. Marks, MD | Michael Shelanski, MD, PhD | ||
 |  | ||
| Andrew Teich, MD, PhD | Ottavio Arancio, MD, PhD |
The "calcium hypothesis" of Alzheimer's disease (AD) has been around for many years, and focuses on the pathological role of elevated intracellular calcium that is reproducibly demonstrated in a variety of AD animal models. Intracellular calcium is a signal that mediates a wide-range of processes, including learning and memory, and several groups have proposed that abnormally elevated calcium levels in AD may contribute to the observed impairment in learning and memory. Elevated intracellular calcium in AD has been proposed to come from both outside the cell as well as from the endoplasmic reticulum (ER). This latter source of calcium enters the cytoplasm in part through ryanodine (RyR) receptors. Although RyR dysfunction has been previously reported in AD animal models, the molecular mechanism of RyR dysfunction has remained largely unexplained until now.
Published this month in Acta Neuropathologica, the laboratory of Dr. Andrew Marks, in collaboration with Drs. Michael Shelanski, Andrew Teich and Ottavio Arancio from the Taub Institute, has taken a major step towards a better understanding of RyR dysfunction in AD. In this study, Marks and colleagues show that RyR2 channels (the major RyR isoform in brain) undergo post-translational remodeling characterized by PKA phosphorylation, oxidation/nitrosylation, and depletion of the channel stabilizing subunit calstabin2 (FKBP12.6) in human sporadic AD, as well as in two well established mouse models of AD (APP+/−/PS1+/− and 3 × Tg-AD). The authors then demonstrate that these post-translational modifications to the RyR2 channels are linked to increased calcium leak from the ER, which mechanistically links these findings to the elevated intracellular calcium levels that are postulated to cause learning and memory impairment in AD.
Are these findings therapeutically relevant? Marks and colleagues perform two separate rescue experiments that suggest they are. First, Marks and colleagues fixed the leaky RyR2 channels in APP+/−/PS1+/− and 3 × Tg-AD mice by orally administering Rycal S107, which is a compound that prevents stress-induced dissociation of calstabin2 from the RyR2 complex. This stabilization of the RyR2 complex inhibited calpain activity and AMPK-dependent tau phosphorylation, and improved hippocampal plasticity and cognitive function. Remarkably, this intervention also reduced amyloid plaque pathology. As a follow-up to this study, Marks and colleagues then crossed APP+/−/PS1+/− mice with RyR2-S2808A+/+ knock-in mice, which have RyR2 channels that are resistant to PKA phosphorylation. This crossing resulted in mice with improved cognition as well as reduced amyloid plaques. Finally, Marks and colleagues investigated the physiologic consequences of a constitutively active RyR2 channel. Mice harboring constitutively leaky PKA phosphomimetic RyR2-S2808D+/+ channels exhibited cognitive and synaptic abnormalities at an early age, consistent with the observation that abnormal RyR2 function may contribute to synaptic and cognitive impairment in AD.
In summary, this study demonstrates a mechanistic link between post-translational modification of RyR2 channels and cognitive dysfunction in AD. Importantly, the therapeutic implications of this finding are nicely demonstrated, and will hopefully spurn new therapies aimed at alleviating ER calcium leak and associated learning and memory abnormalities in AD patients.
Andrew R. Marks, MD
Chairman, Department of Physiology
Founding Director, Wu Center for Molecular Cardiology
Professor, Physiology and Cellular Biophysics; Medicine
arm42@cumc.columbia.edu
Michael Shelanski, MD, PhD
Professor of Pathology & Cell Biology
Henry Taub Professor of Alzheimer's Disease Research
Co-Director, Taub Institute for Research on Alzheimer's Disease and the Aging Brain
oa1@cumc.columbia.edu
Ottavio Arancio, MD, PhD
Professor of Pathology and Cell Biology (in the Taub Institute)
oa1@cumc.columbia.edu
Andrew Teich, MD, PhD
Assistant Professor of Pathology & Cell Biology
aft25@cumc.columbia.edu
Neuropathologic Features of TOMM40 '523 Variant on Late-Life Cognitive Decline

Philip De Jager, MD, PhD, MMSc
In the past 5 years, there has been a tremendous growth in our understanding of the role of genetic variation in the onset of Alzheimer’s disease (AD) and age-related cognitive decline. However, much of this genetic architecture remains to be found and even well-known regions of the human genome can harbor some surprises. Apolipoprotein E (APOE) is the best-known susceptibility gene for late-onset Alzheimer disease (AD). The APOE locus has long been suspected to harbor a non-APOE variant that confers susceptibility to aging-related pathologies and cognitive decline. Recent work has focused on the TOMM40 '523 variant of different lengths that is found near the APOE gene, and the present manuscript, now published online ahead of print in Alzheimer's & Dementia, elaborates its role. Here, new Taub faculty member Dr. Phil De Jager, first author Dr. Lei Yu from Rush Alzheimer's Disease Center, and colleagues examine the role of common neuropathologies in the relationship between TOMM40 '523 genotype and longitudinal cognitive decline, leveraging cognitive, genetic, and neuropathologic data from a large number of community-based older Caucasian Americans who were followed annually for up to 21 years and had undergone brain autopsy after death.
To avoid the effect of the disease-related APOE ε2 and ε4 variants, Dr. De Jager and colleagues focused their analysis on the ε3/3 homozygotes: using ε3/3 homozygotes with at least one copy of the '523 very long (VL) genotype as a reference, both '523 long (L) carriers and '523-short/ short (S/S) genotype had faster cognitive decline. The association of '523-L with cognitive decline was attenuated and no longer significant after controlling for AD-related pathologies, suggesting that enhanced pathology may be the mechanism for this variant. By contrast, the association of '523-S/S was unchanged, which suggests that its mechanism may be distinct. Thus, Yu et al. elaborate the role and mechanism of the TOMM40 susceptibility allele, independent of the role of the well-known APOE- ε2 and ε4 haplotypes, demonstrating that there are two distinct TOMM40 '523 signals in relation to late-life cognitive decline: one signal primarily acts through AD and other common neuropathologies, whereas the other operates through a different mechanism.
Overall, this manuscript is one more step on the path to understanding how variation in the human genome contributes to making human subjects susceptible to developing AD with advancing age. In most individuals with AD, it is the combination of many variants like TOMM40 '523 with environmental risk factors that lead to the accumulation of brain pathology, loss of cognitive function, and, ultimately, a diagnosis of dementia.
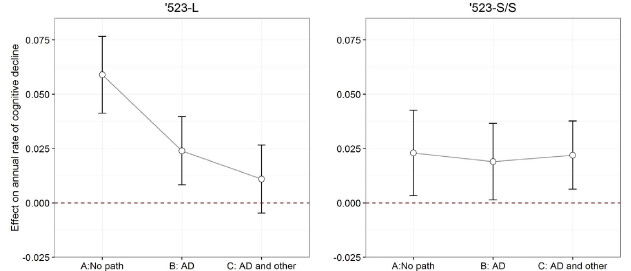 Figure 2: The effects of TOMM40 '523 genotypes on annual rate of cognitive decline before and after the adjustment for neuropathologies. See full caption in manuscript. |
Philip De Jager, MD, PhD, MMSc
Professor of Neurology (in the Taub Institute, the Precision Medicine Initiative, and the Center for Translational and Computational Neuro-immunology)
pld2115@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.